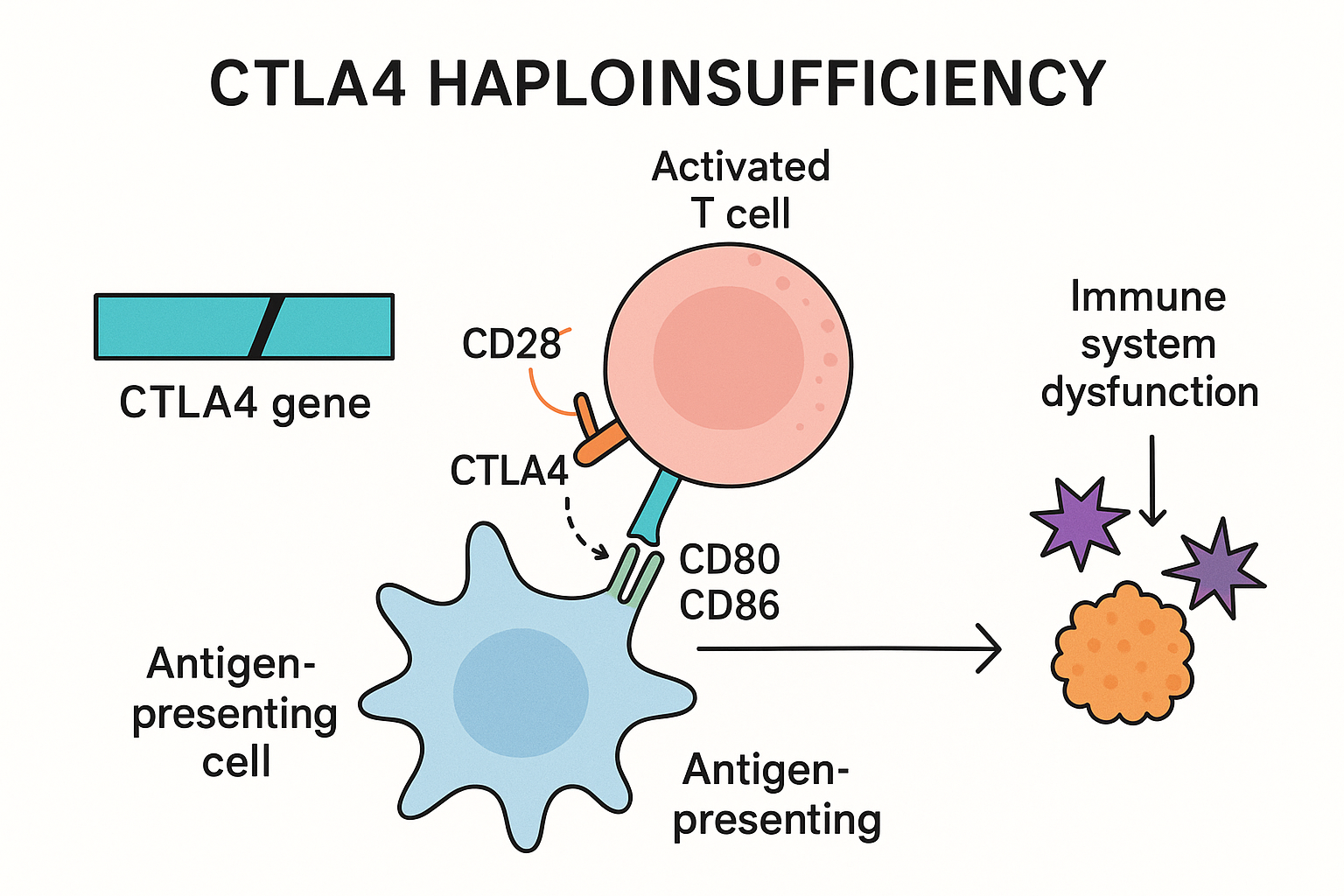
Gambar menunjukkan interaksi sel T dan sel perantara antigen yang mengalami masalah akibat kekurangan separa CTLA 4



Tajuk: Apa itu Penyakit Kekurangan Separa CTLA4
Penyakit imunodefisensi primer (PID) selalunya merujuk kepada penyakit yang diakibatkan oleh satu atau lebih gangguan sistem imun, menjadikan seorang indivividu rentan kepada jangkitan kuman. Jangkitan kuman yang berulang, autoimuniti, limfoproliferasi, proses granulomatus, atopik dan kanser adalah ciri –ciri penghidap PID yang kini diiktiraf sebagai kumpulan penyakit pelbagai dengan keabnormalan sistem imun. Manifestasi klinikal secara keseluruhan bergantung kepada jenis kekurangan imunologi yang berlaku. Jenis jangkitan yang dialami juga boleh berbeza mengikut kategori PID. Antara pelbagai jenis PID yang telah dikenalpasti, kekurangan separa CTLA-4 merupakan salah satu subjenis yang jarang ditemui tetapi mempunyai kepentingan klinikal yang signifikan.
Penyakit Kekurangan Separa CTLA4, juga dikenali sebagai defisensi CTLA 4 atau penyakit CHAI (CTLA-4 haploinsufficiency with autoimmune infiltration), adalah satu gangguan genetik yang jarang berlaku yang disebabkan oleh mutasi pada gen CTLA4, di mana satu salinan gen yang berfungsi tidak ada. Keadaan ini mengganggu fungsi pengawalan normal sistem imun kerana satu salinan gen CTLA4 tidak mencukupi untuk ekspresi gen yang betul.
Akibatnya, individu yang mengalami gangguan ini sering menghadapi disregulasi imun yang dicirikan oleh autoimuniti, hipogamaglobulinemia (paras antibodi yang rendah), jangkitan kuman berulang, dan limfoproliferasi, iaitu keadaan di mana sel imun yang berlebihan menyusup ke dalam organ yang biasanya tidak mengandungi jumlah besar limfosit.
Manifestasi klinikal boleh termasuk penyakit usus, jangkitan pernafasan, serta pembesaran nodus limfa, hati, dan limpa. Selain itu, pesakit juga mungkin mempunyai risiko yang lebih tinggi untuk menghidap kanser limfoma. Diagnosis biasanya dibuat melalui gabungan ujian genetik, keputusan makmal, dan penilaian klinikal.
Pewarisan Kekurangan Separa CTLA-4
Variasi patogenik pada satu salinan gen CTLA4 menyebabkan keadaan genetik yang dikenali sebagai kekurangan separa CTLA-4. Protein CTLA-4, yang penting dalam mengawal aktiviti sistem imun, dihasilkan dan berfungsi dengan kurang baik akibat variasi ini. Gangguan ini diwarisi secara autosom dominan, di mana satu salinan gen yang bermutasi sudah memadai untuk menyebabkan penyakit. Setiap anak mempunyai peluang sebanyak 50% untuk mewarisi mutasi CTLA4 jika salah seorang ibu bapa mempunyai mutasi tersebut. Kebarangkalian seorang anak mewarisi mutasi ini tidak dipengaruhi oleh sama ada adik-beradik mereka turut mewarisi mutasi tersebut atau tidak dan peluangnya adalah sama bagi setiap anak, tanpa mengira jantina adik-beradik yang lain.
Walaupun sesetengah individu yang mewarisi mutasi CTLA4 mungkin menunjukkan tanda-tanda penyakit, ada juga yang tidak mengalami sebarang gejala. Fenomena ini dikenali sebagai penetrasi berubah-ubah (variable penetrance), di mana tidak semua orang yang mempunyai mutasi genetik tersebut menunjukkan simptom klinikal penyakit. Disebabkan oleh keadaan ini, beberapa individu dalam satu keluarga mungkin mempunyai variasi CTLA4, tetapi hanya sebilangan kecil daripada mereka yang menunjukkan simptom, manakala yang lain kelihatan sihat. Adalah penting bagi setiap individu yang didiagnosis mempunyai mutasi CTLA4 untuk menjalani kaunseling genetik, walaupun sesetengah pembawa mutasi tidak menunjukkan sebarang gejala. Pemantauan dan pengurusan awal boleh membantu kerana tanda-tanda klinikal mungkin muncul kemudian dalam hidup. Individu yang terjejas biasanya ditemui dalam beberapa generasi pada sebelah keluarga yang mempunyai mutasi tersebut, memandangkan penyakit ini diwarisi secara autosom dominan.
Apakah simptom kekurangan separa CTLA-4?
Kekurangan separa CTLA-4 dicirikan oleh pengaktifan imunologi yang tidak terkawal, keradangan, dan sering disertai oleh imunodefisensi. Imunodefisensi ini mempunyai persamaan dengan imunodefisensi gabungan pelbagai (CVID) kerana terdapat hipogamaglobulinemia (paras antibodi yang rendah) dan tindak balas antibodi yang terganggu. Malah, sesetengah individu dengan keadaan ini pada awalnya didiagnosis sebagai menghidap CVID. Seperti pesakit CVID, mereka mudah mengalami jangkitan berulang pada sinus dan paru-paru. Jangkitan ini biasanya bermula sejak kanak-kanak dan boleh menjadi lebih teruk apabila usia meningkat. Jangkitan pernafasan berulang adalah antara simptom utama kekurangan CTLA-4. Episod berulang jangkitan bakteria atau virus seperti sinusitis, bronkitis, dan pneumonia selalu berlaku di dalam kalangan individu yang mempunyai kawalan imun yang terganggu.
Penyakit autoimun berlaku apabila sistem imun secara silap menghasilkan antibodi yang menyerang tisu dan sel tubuh sendiri. Sitopenia autoimun, yang didefinisikan sebagai jumlah sel darah putih, sel darah merah, atau platelet yang sangat rendah, merupakan salah satu simptom yang paling kerap berlaku. Autoantibodi boleh menyebabkan kerosakan atau fungsi tisu atau organ tertentu menjadi terganggu, termasuk kelenjar tiroid, pankreas, dan sendi, selain melibatkan sistem hematologi. Beberapa penyakit yang mungkin muncul secara klinikal akibat reaksi imun ini termasuklah alopecia areata, vitiligo, psoriasis, rheumatoid arthritis, gangguan tiroid autoimun, dan diabetes mellitus jenis 1. Bagi sesetengah individu, sistem imun juga boleh menghasilkan kelompok limfosit di dalam organ-organ penting, yang boleh menyebabkan keradangan setempat dan mungkin mengakibatkan kegagalan atau gangguan fungsi organ tersebut. Komplikasi dermatologi seperti psoriasis, artritis keradangan, dan tiroiditis autoimun adalah penyakit autoimun yang kerap berlaku. Selain itu, terdapat juga penyakit autoimun hematologi seperti anemia hemolitik autoimun (AIHA) dan trombositopenia imun (ITP).
Enteropati, atau penyakit usus, sering dicirikan oleh penurunan berat badan, ketidakseimbangan elektrolit terutamanya kehilangan kalium, cirit-birit kronik, dan masalah penyerapan zat makanan. Kehilangan kronik dalam sistem gastrousus boleh memberi kesan yang ketara terhadap pemakanan dan kesihatan keseluruhan. Limfoproliferasi menyebabkan pembesaran limpa, hati, dan nodus limfa. Selain itu, sel imun boleh menyusup ke organ seperti otak, paru-paru, buah pinggang, dan sumsum tulang, menyebabkan keradangan granulomatous dan limfositik. Hipogamaglobulinemia ditunjukkan oleh paras imunoglobulin (IgG, IgA, dan kadangkala IgM) yang rendah. Individu yang mengalami keadaan ini lebih mudah terdedah kepada jangkitan, dan keberkesanan vaksin juga berkurangan disebabkan tindak balas antibodi yang terganggu akibat kekurangan imunologi ini. Individu dengan kekurangan separa CTLA-4 mempunyai risiko yang lebih tinggi untuk menghidap kanser dan limfoma.
Diagnosis
Photo By sumber internet
Gambar menunjukkan ujian makmal pengiraan darah penuh (Full blood count)
Diagnosis bagi kekurangan separa CTLA-4 melibatkan gabungan ujian klinikal, imunologi, dan genetik. Ujian makmal asas termasuk kiraan retikulosit, haptoglobin, ujian Coombs langsung, bilirubin, dan LDH untuk menilai tanda-tanda hemolisis. Paras serum imunoglobulin pula diuji untuk mengesan hipogamaglobulinemia, manakala kiraan darah penuh digunakan bagi mengenal pasti sitopenia seperti kekurangan sel darah merah, putih, atau platelet.
Imunofenotaip melalui sitometri aliran (flow cytometry) sering menunjukkan keabnormalan subset sel T, termasuk pengurangan sel T pengawal selia (Tregs), peningkatan sel T penolong folikular (TFH), serta penurunan sel T pengawal selia folikular (TFR). Selain itu, kelainan pada sel B seperti pengurangan sel B memori dan penilaian subset sel pembunuh semula jadi (NK cells) turut dilakukan. Penilaian fungsi sistem imun boleh dilakukan melalui ujian ke atas proliferasi sel T dan penghasilan sitokin.
Bagi pengesahan diagnosis yang tepat, mutasi heterozigot dalam gen CTLA4 perlu dikenalpasti melalui kaedah penjujukan DNA seperti eksom penuh, genom penuh, atau penjujukan sasaran. Mikrosusunan kromosom pula digunakan untuk mengesan penghapusan berskala besar. Ujian tambahan termasuk pengesanan ekspresi protein CTLA-4 pada sel T teraktif dan Tregs, serta analisis histopatologi biopsi yang menunjukkan penyusupan limfoid yang ketara. Penanda biologi seperti CD25 larut dan BAFF yang meningkat juga boleh menjadi penunjuk tambahan. Kesemua maklumat klinikal, imunologi, dan genetik ini digabungkan untuk mengesahkan diagnosis kekurangan separa CTLA-4.
Rawatan

Photo By sumber internet
Gambar menunjukkan ubat suntikan abatacept
Rawatan bagi individu yang menghidap kekurangan separa CTLA-4 bergantung kepada gejala klinikal dan tahap gangguan sistem imun. Terapi penggantian imunoglobulin (Ig) diperlukan bagi pesakit yang mengalami jangkitan berulang akibat kekurangan antibodi. Selain itu, antibiotik profilaktik (ubat yang diberikan untuk mencegah jangkitan) juga boleh digunakan bagi mengurangkan risiko jangkitan dan mencegah kerosakan organ akibat jangkitan yang kerap. Bagi pesakit yang menunjukkan tanda-tanda gangguan imun yang teruk sehingga menjejaskan fungsi organ, rawatan imunosupresif perlu diberikan bagi menenangkan sistem imun yang tidak terkawal.
Salah satu rawatan yang terbaru ialah abatacept, sejenis ubat suntikan yang telah diluluskan oleh FDA untuk rawatan rheumatoid arthritis dan beberapa jenis psoriasis. Ubat ini berfungsi sebagai pengganti CTLA-4 dan telah menunjukkan hasil yang menggalakkan dalam menangani gangguan imun dalam kalangan pesakit kekurangan CTLA-4. Steroid sistemik seperti prednisone dan methylprednisolone, serta imunosupresan lain seperti sirolimus dan rituximab, turut digunakan secara meluas. Sirolimus berfungsi menghalang pengaktifan sel T dan B dan seterusnya menangani gejala limfoproliferasi yang dialami oleh pesakit kekurangan separa CTLA4. Rituximab pula merupakan antibodi monoklonal yang menyasarkan sel B dan digunakan untuk merawat beberapa jenis kanser dan penyakit autoimun.
Pemindahan sel stem hematopoietik (HSCT) merupakan satu lagi pilihan rawatan bagi kekurangan CTLA-4, namun rawatan ini melibatkan risiko yang tinggi, sebagaimana yang dilihat dalam banyak penyakit imunodefisiensi primer lain.
Penulis:
1. Izazi Binti Arbain
Pelajar Tahun 4, Fakulti Sains Gunaan, Universiti Teknologi Mara Cawangan Perlis, Kampus Arau
Penulis koresponden
2. Prof Madya Dr Intan Juliana Binti Abd Hamid (MD, MMED (Paeds) USM, PhD (Newcastle, UK)
Kumpulan Penyelidikan Penyakit Imunodefisensi Primer, Jabatan Perubatan Klinikal, Institut Perubatan dan Pergigian Termaju, Universiti Sains Malaysia, Kepala Batas, Pulau Pinang.
intanj@usm.my









